

 Previous page
Previous page
Numerically exact spectral lineshape simulations of MAS NMR spectra of extended dipolar coupled spin systems (S. Dusold and A. Sebald)
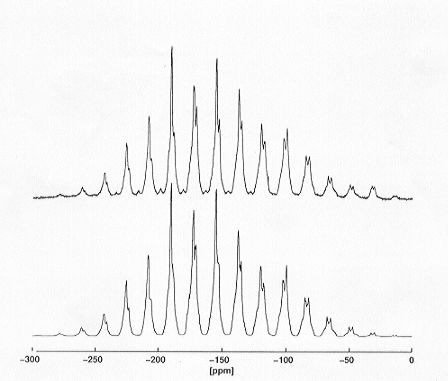
Spectral lineshapes of MAS NMR spectra of dipolar coupled spin-1/2 systems In (n > 1) depend on magnitudes and orientations of all interaction tensors in the spin system. In principle, for coupled spin systems In (n > 2) all geometric information necessary for the complete determination of the corresponding three-dimensional molecular structure is encoded in the lineshapes of straightforward MAS NMR spectra, obtained from polycrystalline powder samples. Numerically exact spectral lineshape simulations in conjunction with iterative fitting procedures are necessary to retrieve this structural information from MAS NMR spectra. In order to permit meaningful and unambiguous determination of molecular geometry from straightforward MAS NMR spectra, suitable lineshape simulation procedures must be both numerically exact and efficient. Speed of computation is a crucial issue, and all steps of the calculations, ranging from smart powder averaging schemes to the evaluation of the periodically time-dependent Hamilton operator and efficient matrix diagonalization methods, need to be optimised. Currently, state-of-the-art computational methods require 2-3 minutes for a full calculation of a four-spin system (in comparison to several hours of computing time necessary only a year ago), using standard computing equipment. Standard MAS NMR experiments in conjunction with iterative lineshape fitting procedures for (currently) up to four-spin systems have now become feasible for purposes of complete structure determination. This is illustrated in Fig. 3.8-13 for a fully 13C-enriched model compound of known single-crystal X-ray structure, the mono-ammonium salt of maleic acid, where the complete geometry of the maleate anion can be determined from iterative lineshape fitting of experimental 13C MAS NMR spectra.
MAS NMR of single crystals (X. Helluy, J. Kümmerlen, A. Sebald, in collaboration with O. Zogal/Wroclaw)
At first glance, a combination of MAS NMR with single crystal samples
seems counterintuitive: traditionally, MAS NMR is the domain of polycrystalline
powder samples, and single-crystal NMR usually operates with stationary
oriented single crystals. Performing MAS NMR experiments on single crystals,
however, offers some advantages. Provided the orientation of the crystal
in the MAS rotor is known, one can take full advantage of the much higher
spectral resolution obtained under MAS conditions. Only a few MAS NMR experiments
are needed to acquire all the information one would otherwise have to extract
from very time-consuming experiments on stationary single-crystal samples,
which in
 |
addition also require special hardware. The high spectral resolution
in single-crystal MAS NMR experiments makes this approach ideally suited
for in-depth investigations of structural phase transitions, amongst other
possible applications. We are using single-crystal 31P MAS NMR
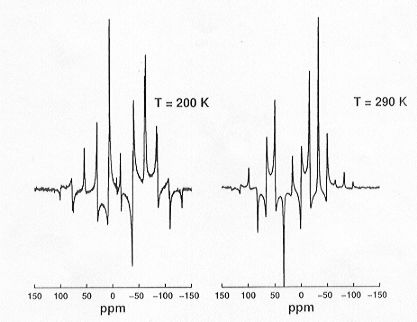
techniques for the study of phase transition properties of glycinium phosphite.
This material is known to undergo a phase transition from the high-temperature
phase to a ferroelectric low-temperature phase at T = 224 K. The analogous
fully deuterated compound shows identical phase transitions, though at
much higher temperatures, and exists in the ferroelectric low-temperature
phase at room temperature. Single crystals of glycinium phosphite survive
the phase transition under MAS conditions and yield high-quality spectra
for both phases (see Fig. 3.8-14). By inspection alone it is already possible
to tell that in the low-temperature phase there are two crystallographically
independent P sites in the unit cell, while the crystallographic symmetry
of the high-temperature phase is higher.
 |
Indirect detection of hydrogen disorder in solid YH2.0± x phases by 89Y CP/MAS NMR (X. Helluy, J. Kümmerlen and A. Sebald, in collaboration with O. Zogal/Wroclaw)
Rare-earth metal hydride phases MxHy display a
wealth of interesting temperature- and composition-dependent material properties.
Amongst these, the yttrium hydride system YHx, which exists
for a wide range of compositions (0<x<3), exhibits dynamic hydrogen
disorder near the YH2 composition. 89Y is a 100 percent
naturally abundant spin-1/2 isotope. In principle, 89Y CP/MAS
NMR experiments at variable temperatures thus permit indirect observation
of order-disorder phenomena in the hydrogen sublattice. In practice, 89Y
CP/MAS NMR experiments are always technically demanding, owing to the very
low Larmor frequency of the 89Y isotope. Within this context,
YH2 phases present additional difficulties for 89Y
CP/MAS NMR experiments as they are metallic. Nevertheless, by taking appropriate
precautions (such as restricting the sample size to a small portion of
the rotor volume, and packing the sample very tightly), we have been able
to spin these samples and tune the rf-circuit of the probe, in the temperature
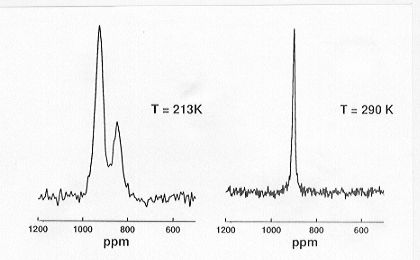
range T = 210 � 290 K. Variable-temperature 89Y CP/MAS NMR spectra
(see Fig. 3.8-15) quantitatively depict the dynamic hydrogen disorder in
these phases. At low temperatures ordering of the hydrogen atoms on tetrahedral
and octahedral sublattice sites occurs, leading to different Y-H coordination
environments and the occurrence of additional 89Y resonances.
Even if 89Y CP/MAS NMR experiments on metallic yttrium hydride
phases are not straightforward experimentally, MAS NMR is a much less expensive
approach than neutron scattering for the investigation of dynamic hydrogen-disorder
effects.
 |
Development of NMR techniques for the determination of molecular conformations (M. Bechmann, S. Dusold and A. Sebald, in collaboration with M. Stumber, U. Haeberlen/Heidelberg, H. Förster/Karlsruhe, M.H. Levitt/Stockholm, T. Lis/Wroclaw and J.N. Evans/Washington)
Molecular conformations play a crucial role in the determination of reaction mechanisms, ranging from biochemical reactions or structure-function relationships of pharmaceuticals to large-scale industrial catalytic synthetic processes. Questions about molecular conformations typically arise for materials which cannot be crystallized (such as membrane proteins). Hence, NMR experiments designed to unambiguously determine torsion angles rather than internuclear distances in molecular target fragments need to be developed and tested. From the view point of solid-state NMR, one of several possible strategies to determine molecular torsional angles relies on the determination of chemical shielding tensor orientations in the molecular frame. The phosphoenol-pyruvate moiety (PEP), a small molecular unit containing one phosphorus and three different carbon atoms, is an important biochemical target for such study. PEP is an "energy-storage" molecule and an active participant in many energy-consuming biochemical reactions, such as synthesis of ATP from ADP.
An extended joint project on this topic has started in several laboratories
around the world. Studies range from synthesis of isotopically labelled
(model) PEP compounds, single-crystal 31P NMR studies and 1H-31P-13C
triple-resonance CP/MAS experiments on model PEP salts to the development
of (double-quantum) 13C MAS NMR experiments, suitable for application
to
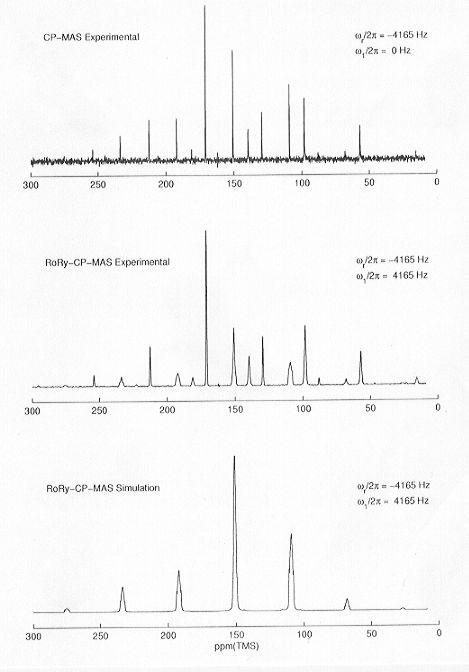 |
"real" biological samples. The first stages of this project are devoted to the determination of 13C and 31P chemical shielding tensor orientations in suitable crystalline model PEP salts, and to the development and implementation of new 13C double-quantum MAS NMR experiments. One of the tasks in our laboratory is the implementation and analysis of 1H-31P-13C triple-resonance CP/MAS NMR experiments for the determination of chemical shielding tensor orientations in model PEP salts with 13C in (low) natural abundance. For this purpose we have chosen so-called 31P-13C rotary resonance recoupling experiments, combined with iterative lineshape fitting methods. Preliminary results and spectral lineshape simulations are shown in Fig. 3.8-16.

Tel: +49-(0) 921 55 3700 / 3766, Fax: +49-(0) 921 55 3769, E-mail: bayerisches.geoinstitut(at)uni-bayreuth.de